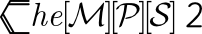 |
CheMPS2
|
|
CheMPS2
|
#include <DMRG.h>
Public Member Functions | |
| DMRG (Problem *Probin, ConvergenceScheme *OptSchemeIn, const bool makechkpt=CheMPS2::DMRG_storeMpsOnDisk, const string tmpfolder=CheMPS2::defaultTMPpath) | |
| Constructor. More... | |
| virtual | ~DMRG () |
| Destructor. | |
| double | Solve () |
| Solver. More... | |
| void | PreSolve () |
| Reconstruct the renormalized operators when you overwrite the matrix elements with Prob->setMxElement() | |
| void | calc2DMandCorrelations () |
| Calculate the 2-RDM and correlations. Afterwards the MPS is again in LLLLLLLC gauge. | |
| void | calc_rdms_and_correlations (const bool do_3rdm, const bool disk_3rdm=false) |
| Calculate the reduced density matrices and correlations. Afterwards the MPS is again in LLLLLLLC gauge. More... | |
| TwoDM * | get2DM () |
| Get the pointer to the 2-RDM. More... | |
| ThreeDM * | get3DM () |
| Get the pointer to the 3-RDM. More... | |
| void | Symm4RDM (double *output, const int ham_orb1, const int ham_orb2, const bool last_case) |
| Obtain the symmetrized 4-RDM terms 0.5 * ( Gamma4_ijkl,pqrt + Gamma4_ijkt,pqrl ) with l and t fixed, after the 3-RDM has been calculated. More... | |
| Correlations * | getCorrelations () |
| Get the pointer to the Correlations. More... | |
| double | getSpecificCoefficient (int *coeff) const |
| Get a specific FCI coefficient. The array coeff contains the occupation numbers of the L Hamiltonian orbitals. It is assumed that the unpaired electrons are all alpha electrons, and that this number equals twice the total targeted spin. More... | |
| double | getFCIcoefficient (int *alpha, int *beta, const bool mpi_chemps2_master_only=true) const |
| Get a specific FCI coefficient. The arrays alpha and beta contain the alpha and beta occupation numbers of the L Hamiltonian orbitals. More... | |
| void | deleteStoredMPS () |
| Call "rm " + CheMPS2::DMRG_MPS_storage_prefix + "*.h5". | |
| void | deleteStoredOperators () |
| Call "rm " + tempfolder + "/" + CheMPS2::DMRG_OPERATOR_storage_prefix + string(thePID) + "*.h5";. | |
| void | activateExcitations (const int maxExcIn) |
| Activate the necessary storage and machinery to handle excitations. More... | |
| void | newExcitation (const double EshiftIn) |
| Push back current calculation and set everything up to calculate a (new) excitation. More... | |
Static Public Member Functions | |
| static void | PrintLicense () |
| Print the license. | |
DMRG class.
The DMRG class solves the Problem with its given parameters. A fully SU(2) symmetric MPS wavefunction is variationally optimized in a two-site sweep algorithm. When the solution has been reached, the converged energy and spin contracted 2DMs can be accessed. For more information, please take a look at
S. Wouters, W. Poelmans, P.W. Ayers and D. Van Neck,
CheMPS2: a free open-source spin-adapted implementation of the density matrix renormalization group for ab initio quantum chemistry,
Computer Physics Communications 185, 1501-1514 (2014)
http://dx.doi.org/10.1016/j.cpc.2014.01.019
http://arxiv.org/abs/1312.2415
S. Wouters and D. Van Neck,
The density matrix renormalization group for ab initio quantum chemistry,
European Physical Journal D 68, 272 (2014)
http://dx.doi.org/10.1140/epjd/e2014-50500-1
http://arxiv.org/abs/1407.2040
The user manual: http://sebwouters.github.io/CheMPS2/index.html
| CheMPS2::DMRG::DMRG | ( | Problem * | Probin, |
| ConvergenceScheme * | OptSchemeIn, | ||
| const bool | makechkpt = CheMPS2::DMRG_storeMpsOnDisk, |
||
| const string | tmpfolder = CheMPS2::defaultTMPpath |
||
| ) |
Constructor.
| Probin | The problem to be solved |
| OptSchemeIn | The optimization scheme for the DMRG sweeps |
| makechkpt | Whether or not to save MPS checkpoints in the working directory |
| tmpfolder | Temporary folder on a large partition to store the renormalized operators on disk (by default "/tmp") |
Definition at line 36 of file DMRG.cpp.
 Here is the call graph for this function:
Here is the call graph for this function:| void CheMPS2::DMRG::activateExcitations | ( | const int | maxExcIn | ) |
| void CheMPS2::DMRG::calc_rdms_and_correlations | ( | const bool | do_3rdm, |
| const bool | disk_3rdm = false |
||
| ) |
Calculate the reduced density matrices and correlations. Afterwards the MPS is again in LLLLLLLC gauge.
| do_3rdm | Whether or not to calculate the 3-RDM |
| disk_3rdm | Whether or not to use disk in order to avoid storing the full 3-RDM of size L^6 |
Definition at line 40 of file DMRGtechnics.cpp.
Here is the call graph for this function: Here is the caller graph for this function:
|
inline |
|
inline |
|
inline |
Get the pointer to the Correlations.
Definition at line 128 of file DMRG.h.
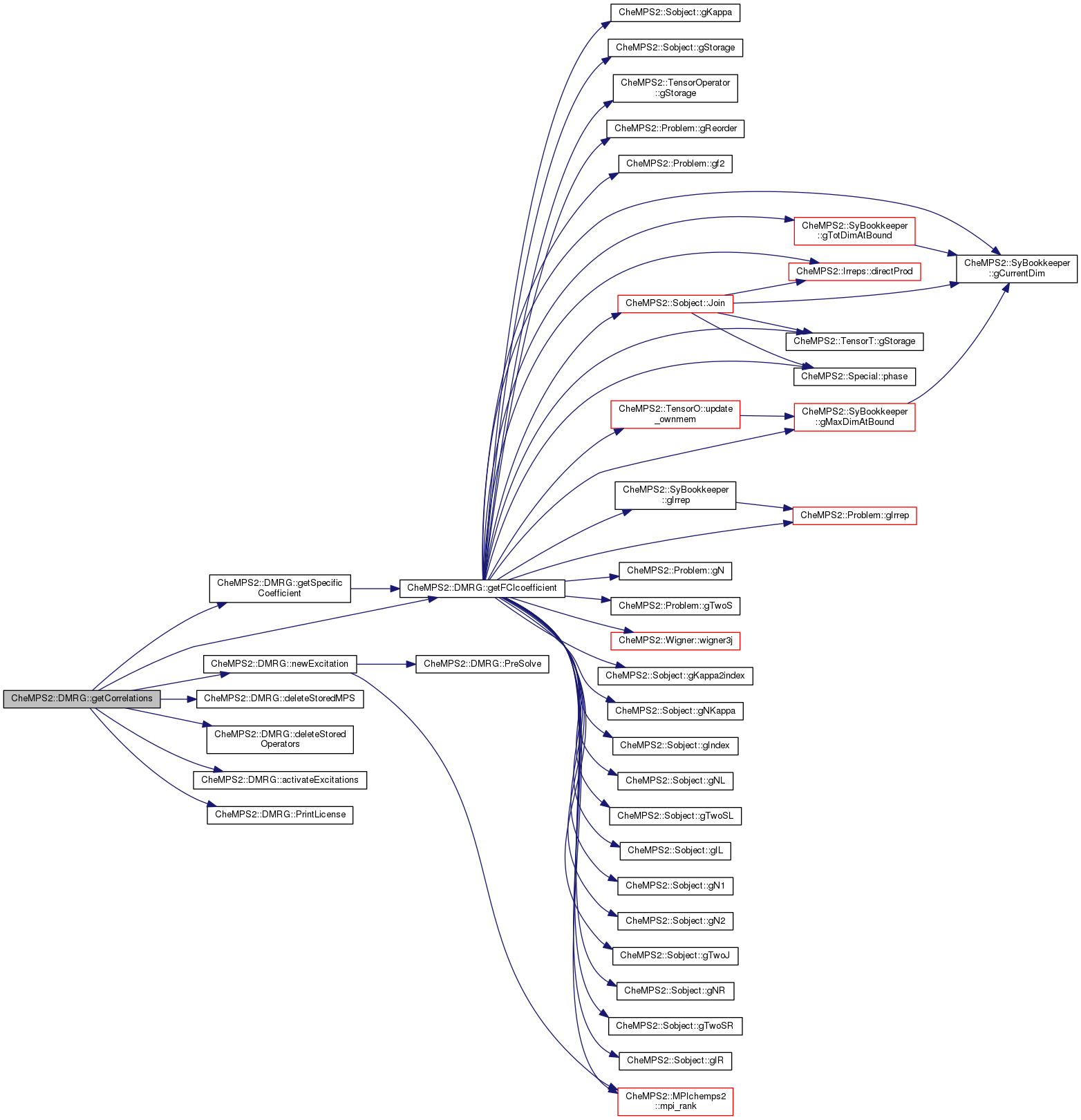
Here is the call graph for this function: Here is the caller graph for this function:| double CheMPS2::DMRG::getFCIcoefficient | ( | int * | alpha, |
| int * | beta, | ||
| const bool | mpi_chemps2_master_only = true |
||
| ) | const |
Get a specific FCI coefficient. The arrays alpha and beta contain the alpha and beta occupation numbers of the L Hamiltonian orbitals.
| alpha | Array containing the alpha electron occupation numbers of the L Hamiltonian orbitals (occupations can be 0 or 1). |
| beta | Array containing the beta electron occupation numbers of the L Hamiltonian orbitals (occupations can be 0 or 1). |
| mpi_chemps2_master_only | When running with MPI, whether only the master process should calculate the FCI coefficient. If false, any process can calculate the FCI coefficient, and it won't be broadcasted (allows for parallel FCI coefficient calculation). |
Definition at line 341 of file DMRGtechnics.cpp.
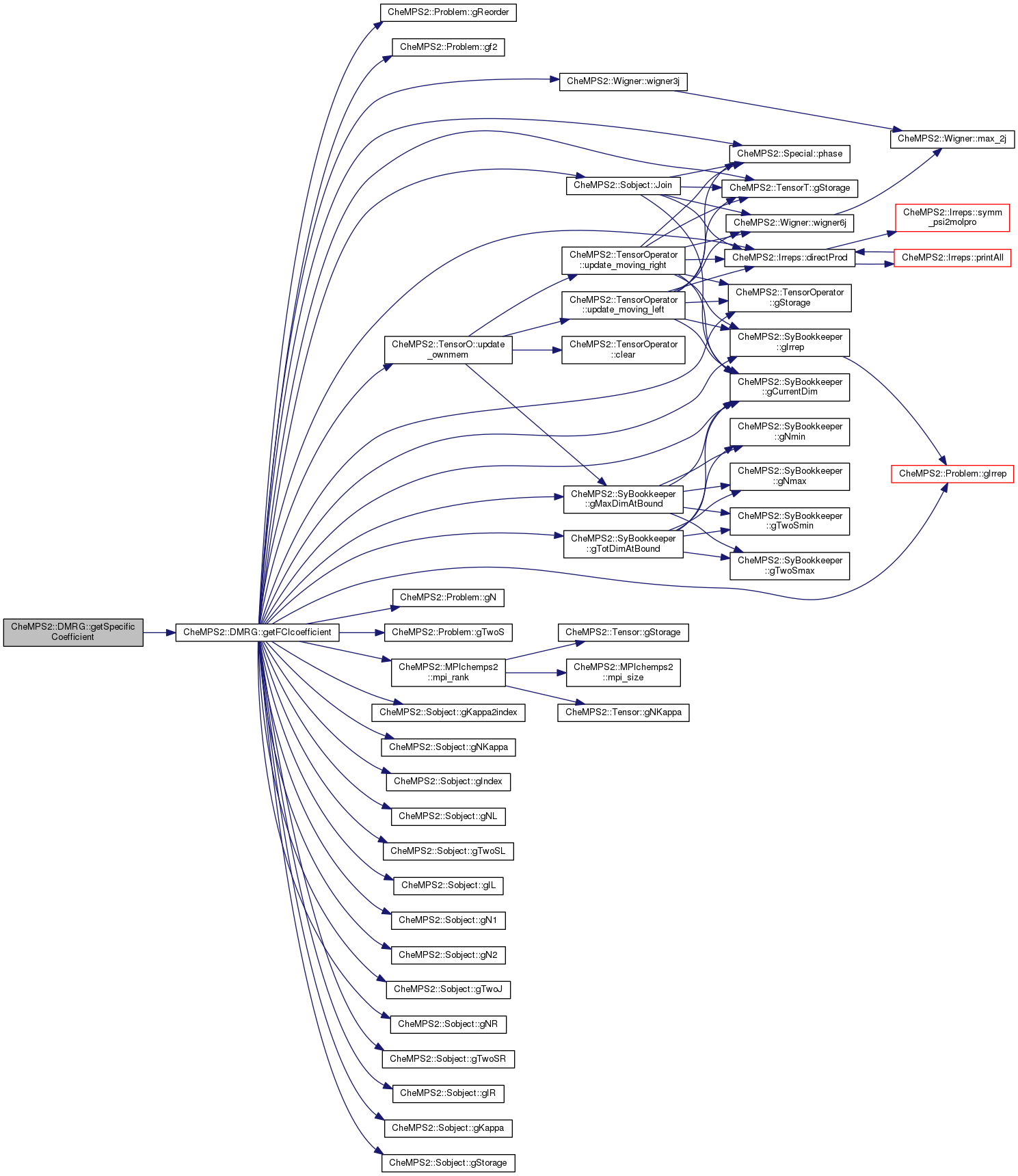
Here is the call graph for this function: Here is the caller graph for this function:| double CheMPS2::DMRG::getSpecificCoefficient | ( | int * | coeff | ) | const |
Get a specific FCI coefficient. The array coeff contains the occupation numbers of the L Hamiltonian orbitals. It is assumed that the unpaired electrons are all alpha electrons, and that this number equals twice the total targeted spin.
| coeff | Array containing the occupation numbers of the L Hamiltonian orbitals (occupations can be 0, 1, or 2). |
Definition at line 324 of file DMRGtechnics.cpp.
Here is the call graph for this function: Here is the caller graph for this function:| void CheMPS2::DMRG::newExcitation | ( | const double | EshiftIn | ) |
Push back current calculation and set everything up to calculate a (new) excitation.
| EshiftIn | To the Hamiltonian, a level shift is introduced to exclude the previously calculated MPS: Hnew = Hold + EshiftIn * | prev> <prev| |
Definition at line 395 of file DMRG.cpp.
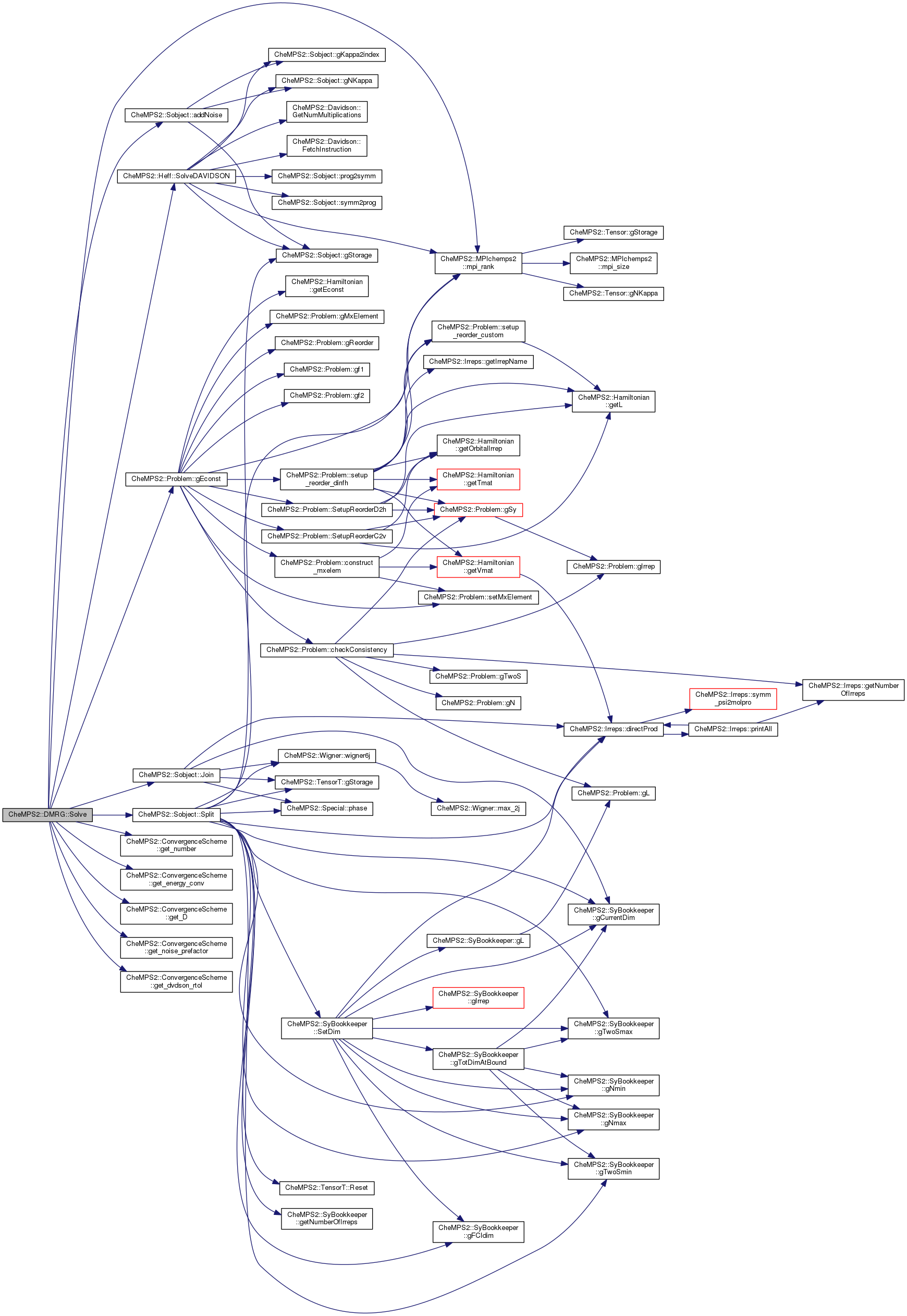
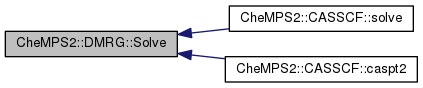
Here is the call graph for this function: Here is the caller graph for this function:| double CheMPS2::DMRG::Solve | ( | ) |
| void CheMPS2::DMRG::Symm4RDM | ( | double * | output, |
| const int | ham_orb1, | ||
| const int | ham_orb2, | ||
| const bool | last_case | ||
| ) |
Obtain the symmetrized 4-RDM terms 0.5 * ( Gamma4_ijkl,pqrt + Gamma4_ijkt,pqrl ) with l and t fixed, after the 3-RDM has been calculated.
| output | Array to store the symmetrized 4-RDM terms in Hamiltonian index notation: output[ i + L * ( j + L * ( k + L * ( p + L * ( q + L * r )))) ] = 0.5 * ( Gamma4_ijkl,pqrt + Gamma4_ijkt,pqrl ). |
| ham_orb1 | The Hamiltonian index of the first fixed orbital. |
| ham_orb2 | The Hamiltonian index of the second fixed orbital. |
| last_case | If true, everything will be set up to allow to continue sweeping. |
Definition at line 41 of file DMRGfock.cpp.
Here is the call graph for this function: Here is the caller graph for this function: 1.8.11
1.8.11